GeomBD
Rigid Body Brownian Dynamics
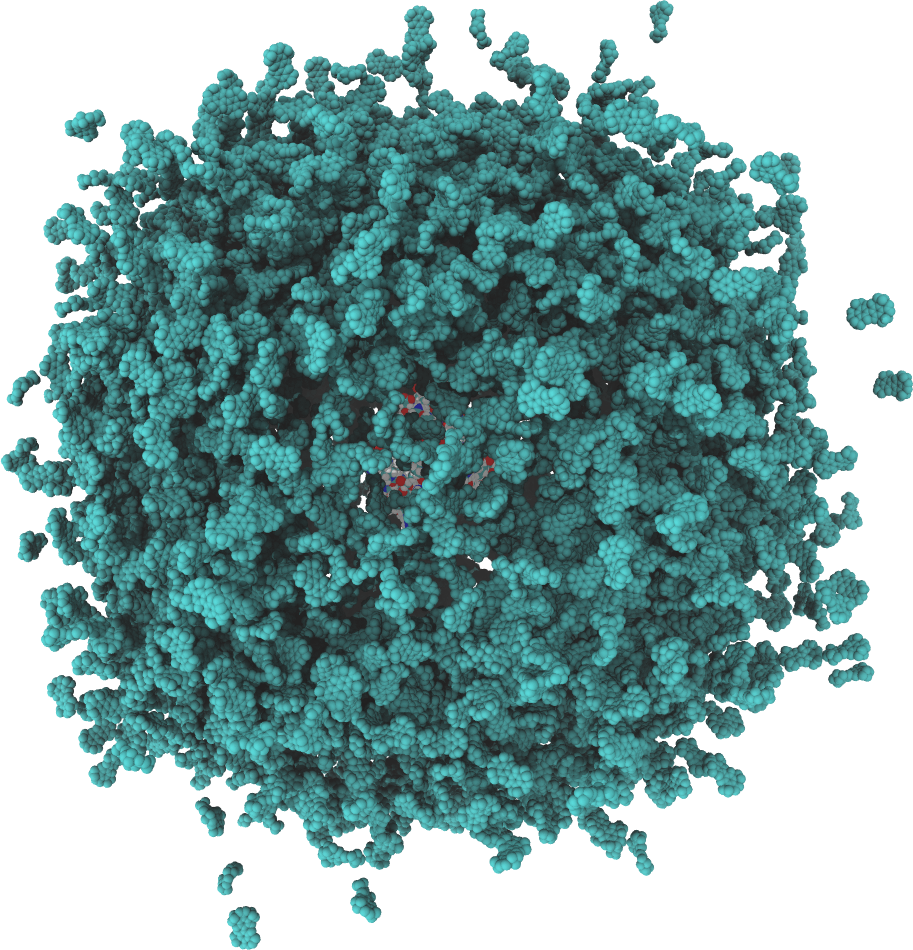
GeomBD is a rigid body Brownian dynamics software for determining molecular association rates in biomolecular or engineered systems. Association of ligands and other molecules to various receptor molecules or surfaces can be investigated under a vast number of system configurations. For example, multi-enzyme complexes, crowded cellular environments, enzyme bioconjugtes, biosensors, and much more can be computationally modeled and simulated to elucidate reaction kinetics and pathways. Any novel, engineered system can be added to the GeomBD parameter files and simulated to investigate the effects of any number of system properties. Tools are provided to determine non-specific ligand-receptor association residence times, visualize common non-specific association sites and binding pathways, calculate diffusion coefficients, and calculate intermolecular interaction strengths.
- GeomBD Version 3 (GeomBD3)
- GeomBD3 beta1 - Stabilized GeomBD version 3
- GitHub - GeomBD3 package and other analysis codes for specific projects/systems
[1] Timothy Cholko, Shivansh Kaushik, Kingsley Wu, and Chia-en A. Chang, GeomBD3: Brownian Dynamics Simulation Software for Biological and Engineered Systems, 2022[link]
[2] Roberts, C. C. and Chang, C-E. A.*, Modeling of Enhanced Catalysis in Multi-enzyme Nanostructures: Effect of Molecular Scaffolds, Spatial Organization, and Concentration. J. of Chemical Theory and Computation, 2015 [link]
[3] Roberts, C. C. and Chang, C-E. A.*, Analysis of Ligand-Receptor Association and Intermediate Transfer Rates in Multi-enzyme Nanostructures with All-Atom Brownian Dynamics Simulations. J. of Physical Chemistry B. 2016 [link]
BKiT: Binding Kinetics Tool
BKiT is a computational approach for investigating drug unbinding kinetics using molecular dynamics simulations and milestoning theory. The underlying strategy is described in Tang, Z.; Chen, S.-H.; Chang, C. A. J. Chem. Theory Comput. 2020, 16, 1882–1895 (bioRxiv 169607). Simulation input files from that paper are available here.
T-Analyst
T-Analyst is a user-friendly computer program for analyzing protein dynamics and conformational changes from MD simulations in internal bond-angle-torsion coordinates. The program is provided with tutorial and full source code in Perl.
[1] Ai, R., Fatmi, Q. and Chang, C-E. A., T-Analyst: a program for efficient analysis of protein conformational changes by torsion angles. J. of Computer-Aided Molecular Design. 2010 [link]
MD_traj for Ab42 coil conformation
by Kingsley Wu
PLpro 1us MD trajectories
(100K frames, no water)by Yuliana Bosken and Timothy Cholko
SARS-CoV-2 6W9C APO
SARS-CoV-2 6WRH APO
SARS-CoV-2 6W9C-3k A pose
SARS-CoV 4OW0 APO
SARS-CoV 4OW0-3k
MD Input Files
Water trajectories are also available upon request.